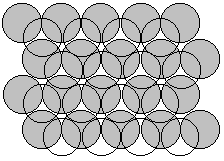
Since ions and many molecules are basically spherical objects, the structures of solids arise from packing of spheres. Generally the most effective way to do this is to put objects as close to each other as possible (closest packed structures). There are two common closest packed structures.
Hexagonally closest packed (hcp): an ABAB sequence of spheres
solid circles are the 1st, 3rd, 5th, etc., layers open circles are the 2nd, 4th, 6th, etc. layers
There are two types of vacancies or holes: those with four neighbors (Td holes) and those with six neighbors (Oh holes).
~74% of the volume is empty; each sphere has twelve nearest neighbors adjacent to it. This is known as the Coordination Number
Cubic closest packed: ABCABC sequence of spheres
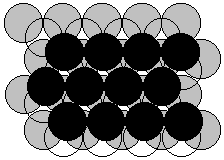
ccp = face centered cubic, i.e. draw a cube and put spheres at the corners and the center of each of the faces
Ionic compounds nearly always form closest packed structures if the ions are simple and spherical (nonspherical ions change things). Normally, the largest ion (typically the anion) forms the lattice and the smaller ion fills the holes - either Td or Oh holes.
Metals and other neutral covalent compounds also often form closest packed structures. Alloys, which have more than one metal atom, can form by the larger atom forming the lattice and the smaller atom filling holes (either Td or Oh)
When closest packed structures are not formed (and even when they are), the usual way of thinking about solid state crystalline structures is to find the smallest unit that repeats itself via translational symmetry over the whole macroscopic structure. This smallest repeat unit is called the unit cell. There are 7 types of unit cells called crystal systems. The atoms or ions in the crystal do not determine the unit cell; it is determined by the translational symmetry!
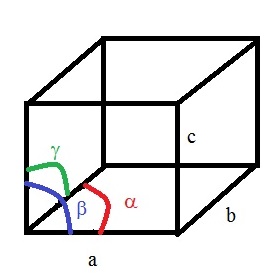
To determine the crystal system, 6 parameters are specified: 3 distances and 3 angles, and the occupation of the cell
The 7 crystal systems are determined by symmetry restrictions, as shown below:
Namelattice parametersDefining parameters Required Symmetries
Triclinica ≠ b ≠ c, α ≠ β ≠ γ ≠ 90° a, b, c, α, β, γNone
Monoclinica ≠ b ≠ c, α = β = 90° ≠ γ ≠ 90° a, b, c, βone C2 and/or σ
Orthorhombica ≠ b ≠ c, α = β = γ = 90° a, b, cThree perpendicular C2 and/or σ
Rhombohedrala = b = c, α = β = γ ≠ 90° a, αone C3
Tetragonala = b ≠ c, α = β = γ = 90° a, cone C4
Hexagonala = b ≠ c, α = β = 90° γ = 120° a, cone C6
Cubica = b= c, α = β = γ = 90° afour C3 with Td relative orientation
The 7 crystal systems can have different occupation of critical points, known as lattice points. The lattice points are the corners of the unit cell, the center of the faces of the unit cell, or in the center of the body of the unit cell
If only the corners are occupied, this is referred to as a Primitive cell.
If the corners and the center lattice points are occupied, this is referred to as a Body-Centered cell.
If the corners and all of the faces are occupied, this is referred to as a Face-Centered cell.
If the corners and two opposing faces are occupied, this is referred to as an End-Centered cell.
Combining the 7 crystal systems with the lattice points gives 14 symmetry allowed structures, known as Bravais Lattices:
Primitive Cubic
Body-Centered Cubic
Face-Centered Cubic
Primitive Tetragonal
Body-Centered Tetragonal
Primitive Orthorhombic
Body-Centered Orthorhombic
End-Centered Orthorhombic
Face-Centered Orthorhombic
Primitive Monoclinic
End-Centered Monoclinic
Triclinic, Rhombohedral, and Hexagonal cells can only be primitive so no modifier is added
Each unit cell can be packed in different ways, depending on the translational symmetry.
In addition to simple translations, there also can be more complex patterns: glide planes (a mirror plus a translation) and screw axes (a C2 plus a translation)
Combining the Bravais Lattices with the symmetry allowed translations leads to 230 combinations, known a Space Groups.
Whenever crystal structures are reported the Space Group, the cell type (which is done using a notation we won't discuss), and the cell parameters (a, b, c, α, β γ as needed) are given.
In real life lattices are not perfect; defects always exist, even at 0 K (this is thermodynamically required: enthalpy prefers perfection but entropy prefers randomness; get a balance with mostly perfect but a few defects).
Schottky defects: equal numbers of cations and anions are missing from the lattice
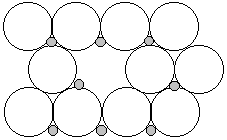
Frenkel defects: small ion (usually the cation) moves to interstitial site creating a hole that doesn't belong or pushing larger ion off the lattice site
Nonstoichiometric Compounds: many compounds do not follow the freshman chemistry idea that stoichiometry of compounds is in the ratio of small whole numbers.
FeO is more typically is found as FexO where x~0.95-0.99
Charge neutrality must still be maintained so a better description is (Fe2+)3x-2(Fe3+)2-2xO
F centers: e– become part of the lattice
NaCl + δNa ⇄ Na1+δ(Cl)(e–)δ