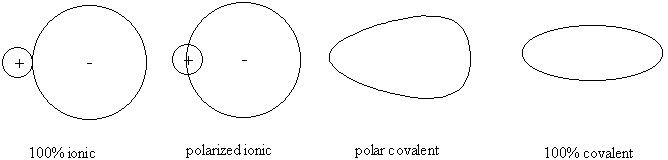
The idea of determining the sizes of ions is challenging. The usual assumption is that a given ion has the same size in any compound. This assumption is partially true.
The problem is that we can easily measure the distance between atomic nuclei, establishing where an anion ends and a cation begins is subjective.
Theoretically, one can calculate electron densities and use the minimum between atomic nuclei as the boundary. This leads to somewhat varying results owing to the different basis sets and computational approaches used.
Experimentally, electron densities can be measured but this requires high quality crystals and better than routine x-ray measurements. This has been done for a few ionic compounds.
Using standard x-ray diffraction techniques, Landé used LiI, a small cation matched with a large anion, and made the assumption that the I– ion was closest packed and that the Li+ ion filled the Oh holes (face-centered cubic structure). With this assumption, ro = r+ + r– and d(I––I–) = 2r–.
Then, assuming r+ and r– are constant from compound to compound, measuring ro for the other group 1 halides led to a reasonably consistent set of radii. However, CsCl was an outlier - it is body-centered cubic. This led to the extension that the coordination number of the ion changes the ionic radius. Further, using the same ideas for multivalent ions led to further discrepancies.
To address this problem, Pauling proposed that ionic radii are proportional to the effective nuclear charge, Z*, which he then used to develop an equation relating multivalent radii to multivalent radii: rZ/r±1 = Z–2/(n – 1) where Z is the formal charge on the ion and n is the Born exponent.
All of these concepts were put together by Shannon (Acta Cryst., 1972, A32, 751) to give a fairly consistent set of ionic radii
Ionic radii can be used to predict structures and lattice energies
The radius ration = γ = rsmall/rlarge
If γ < 0.225, then the small ion will fill Td holes.
If γ 0.225 < 0.414, then the small ion will fill Td holes but push the larger ions apart.
If γ 0.414 < 0.732, then the small ion will fill Oh holes.
If γ > 0.732, then the small ion will be too large for a closest packed structure and the coordination number will be 8.
M+(g) + X–(g) → MX(s) ΔH ~ –Elat
This is a difficult measurement - hard to get enough gas phase ions to achieve big enough heats
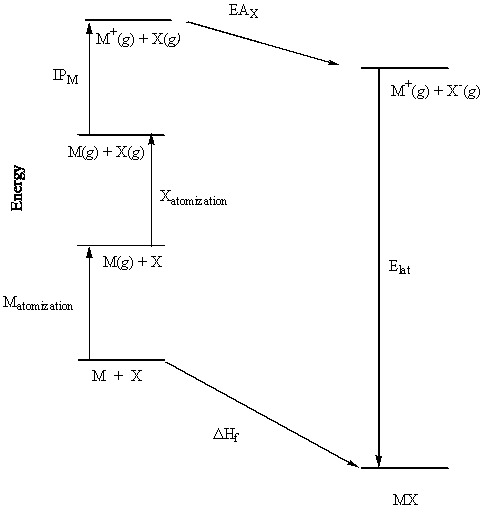
The sum of the known reactions gives the desired reaction; summing the energies gives the lattice energy. Born-Haber cycles must be set up for each compound to account for the correct states of matter, stoichiometry, etc.
Example: NaBr
| Na(s) → Na(g) | ΔHsublimation = +107.8 kJ/mole (M atomization) |
| ½Br2(l) → ½Br2(g) | ½ΔHvaporization = ½(+53.4) = +26.7 kJ/mole |
| ½Br2(g) → Br(g) | ½ΔHdissociation = ½(+190.2) = +95.1 kJ/mole |
| (Xatomization = ½ΔHvaporization + ½ΔHdissociation) | |
| Na(g) → Na+(g) + e– | IP = +495.4 kJ/mole |
| Br(g) + e– → Br–(g) | –EA = –324.6 kJ/mole |
| Na+(g) + Br–(g) → NaBr(s) | –Elat |
| NaBr(s) → Na(s) + ½Br2(l) | –ΔHf = –(–361.4) = 361.4 kJ/mole |
Summing all of these reactions gives nothing so that summing the energies must add to zero:
ΔHsublimation + ½ΔHvaporization + ½ΔHdissociation + IP – EA – Elat – ΔHf = 0
Elat = +107.8 + 26.7 + 95.1 + 495.4 – 324.6 + 361.4 = 761.8 kJ/mole
Compares to 712.4 kJ/mole from Born-Landé equation
With such large lattice energies, why do ionic compounds dissolve?
Consider another Born-Haber cycle:
| Na+(aq) + Cl–(aq) → NaCl(s) | –ΔHsolvation |
| NaCl(s) → Na+(g) + Cl–(g) | Elat |
| Na+(g) → Na+(aq) | Esolv+ |
| Cl–(g) → Cl–(aq) | Esolv– |
So that –ΔHsolvation + Elat + Esolv+ + Esolv– = 0
ΔHsolvation is usually pretty small - a few 10s of kJ/mol; clearly much different from Elat
This says that Esolv+ + Esolv– ~ Elat but with opposite signs
Find Esolv in a manner similar to finding Elat. This gives:
E±solv = –N0(Z±)2e2(1 – 1/ε)/ 8πεor±
r = ionic radius
Z = ionic charge
ε = solvent dielectric constant
+ or – refers to either cation or anion Z or r
1. Cations are smaller than anions so that r is much smaller for cations than for anions. This means that Esolv for cations is much larger than for anions, i.e., solvation is caused primarily by the cations and not the anions.
2. Solvation energy increases with the square of ionic charge.
3. Solvents with high dielectric constants solvate ionic compounds better; a dielectric constant near 1 will give near zero solvation energy (water has ε ~ 87 at room T)
Consider AgCl: has NaCl structure, ro = 2.77 Å (compared to 2.81 Å for NaCl) yet AgCl is only sparingly soluble in water, mp is ~350 oC lower than NaCl (801 vs 455 oC), Lattice energy is Elat = 907.4 kJ/mol (compared to 786.8 kJ/mol for NaCl).
Why so different?
Polarization effects (covalency): AgCl has more covalent character than NaCl so has less classical ionic character. Consider the limits of bonding: the difference between polarized ionic and polar covalent is merely one of degree. Polarization of "ionic" compounds occurs when the cation is very small and the anion is very large and polarizable. This also can occur in transition metal ions where the d shell electrons are more polarizable than s or p electrons. Transition metals are also more polarizing to anions because of a larger Z*.